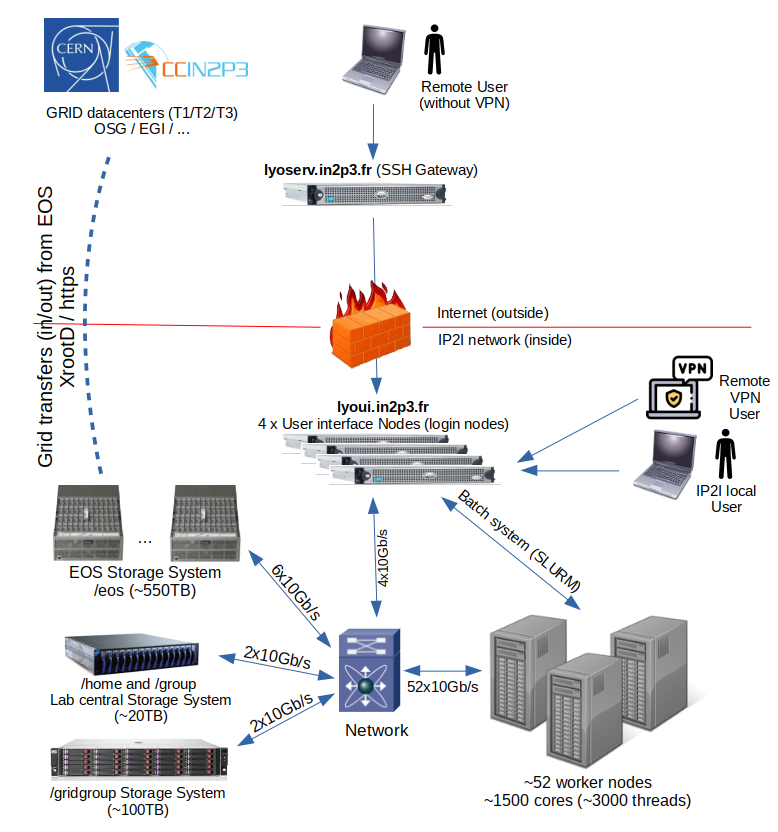
IP2I HTC Farm
A FARM is a group of machines aggregated together dedicated to computing. It is composed of a group of front machines (named UI : "User Interface" of the cluster) and a large number of individual computing machines, called worker nodes, and possibly heterogeneous. Theses machines are managed by a scheduler. The scheduler used is SLURM
What is SLURM ?
SLURM (Simple Linux Utility for Resource Management) is a resource manager and task scheduler for LINUX clusters. It is an open source software used wildly in high performance computing, It allows the best distribution of computing resources (CPU, GPU, RAM) between users by managing queues with priorites between users, between groups and ressources.
A user's processes are confined to the resources (CPUs in particular) that he has reserved to it by the scheduller : he cannot access CPUs reserved by other users. This ensures a certain reproducibility of the executions of the same program, especially in terms of computing times (unlike what happens on machines with free access).
In SLURM, a job corresponds to a request for resource allocation (CPU, RAM, computing time) by a user. A job is composed of steps and each step performs one task or more in parallel. A task corresponds to a process, a task can use one or more CPUs.
Hardware and software
SLURM CPU Farm
The farm currently features 3072 threads :
| Number of workers | Worker Type and names | CPU | RAM / worker | Number of threads / worker |
|---|---|---|---|---|
| 4 | Dell C6420 lyowork[033-036] |
Intel(R) Xeon(R) Gold 6130 CPU @ 2.10GHz | 192 GB | 64 (2 CPUs * 16 cores * 2 threads) |
| 4 | Dell C6320 lyowork[037-040] |
Intel(R) Xeon(R) CPU E5-2680 v4 @ 2.40GHz | 160 GB | 56 (2 CPUs * 14 cores * 2 threads) |
| 52 | Dell C6320 lyowork[001-028,041-064] |
Intel(R) Xeon(R) CPU E5-2680 v3 @ 2.50GHz | 144 GB | 48 (2 CPUs * 12 cores * 2 threads) |
| 1 | Dell R740 GPU lyowork[029] |
Intel Xeon Gold 6248R CPU @ 3.00GHz | 192 GB | 96 (2 CPUs * 24 cores * 2 threads) |
| 61 workers | TOTAL | 8,9 TB | 1536 cores = 3072 threads |
SLURM GPU node
One particular worker node lyowork029 is a GPU server is also available in the gpu partition with 3 * NVIDIA Quadro RTX 6000 GPUs :
- 96 cores : Intel Xeon Gold 6248R CPU @ 3.00GHz (24 cores * 2 threads)
- 192 GB RAM
- Storage : 1.8 TB on SSD in
/scratch - GPU : 3 * NVIDIA Quadro RTX 6000
This node is dedicated to Machine Learning and is described in deph in the page How to use the IP2I GPU server ?
Software configuration
Each node has a common configuration :
- Centos 7
- CVMFS configured (
/cvmfs/alice.cern.ch,/cvmfs/cms.cern.ch,/cvmfs/oasis.opensciencegrid.org,/cvmfs/gwosc.osgstorage.org,/cvmfs/dirac.egi.eu,/cvmfs/grid.cern.ch,/cvmfs/sft.cern.ch,/cvmfs/geant4.cern.ch,/cvmfs/ilc.desy.de,/cvmfs/singularity.opensciencegrid.org,/cvmfs/francegrilles.in2p3.fr,/cvmfs/fermilab.opensciencegrid.org,/cvmfs/dune.opensciencegrid.org. ...) - all grid middleware software installed
- A bunch of software installed
User interface nodes (interactive nodes)
The SLURM farm workers are only accessible from 4 interactive machines: lyoui1 to lyoui4 (you can use the DNS alias lyoui which redirects you to the less busy machine with the command ssh lyoui).
These User interface nodes are not reachable from outside of the IP2I network, if you need to access them from outside, you need to connect first to the IP2I's SSH gateways lyoserv.in2p3.fr : ssh lyoserv.in2p3.fr, then ssh lyoui.
| Number | Name alias | Real server names | CPU | RAM / server | Number of threads / worker |
|---|---|---|---|---|---|
| 4 | lyoui.in2p3.fr |
lyowork[1-4].in2p3.fr |
Intel(R) Xeon(R) CPU E5-2680 v3 @ 2.50GHz | 144 GB | 48 (2 CPUs * 12 cores * 2 threads) |
Some recommendations for the usage of these User interface (UI) nodes :
- You can launch only short CPU intensive processing (few hours) in these interactive machines but not more. If you need to launch heavier CPU intensive processing, use the SLURM batch system for this usage.
- You should not use
/tmpto store temporary files, this is a tiny directory (less than 1GB) only used for system files. Instead, use the/scratchdirectory which is more than 500GB (up to few TB). All files stored in the/scratchdirectory will be automatically deleted after 90 days.
Quick start
Log on one of the 4 interactive machines lyoui1 to lyoui4. These hosts are behind a DNS load balanced alias lyoui.in2p3.fr :
ssh lyoui
The main commands you need to know about to use SLURM are sinfo, srun, sbatch, squeue, scancel :
sinfo: gives information about the partitions (queues) and the worker nodessrun: launch a tasksbatch: launch a job (script)squeue: information about the list of jobs (running, waiting...) in the partitionsscancel: to cancel a jobscontrol show job|partition <jobid|partition_name>: gives some information about a job or a partitionsacct: accounting information about past jobs (State, how much CPU used, how much memory used...)
List the queues (called partitions in SLURM):
sinfo
This command gives something like :
PARTITION AVAIL TIMELIMIT NODES STATE NODELIST
normal* up 1-00:00:00 11 drain lyowork[001-003,037-038,063-068]
normal* up 1-00:00:00 7 mix lyowork[024-026,049,058-059,062]
normal* up 1-00:00:00 45 alloc lyowork[004-023,027-028,033-036,040-048,050-057,060-061]
normal* up 1-00:00:00 1 down lyowork039
test up 1:00:00 11 drain lyowork[001-003,037-038,063-068]
test up 1:00:00 7 mix lyowork[024-026,049,058-059,062]
test up 1:00:00 45 alloc lyowork[004-023,027-028,033-036,040-048,050-057,060-061]
test up 1:00:00 1 idle lyowork029
long up 45-00:00:0 7 drain lyowork[001-003,065-068]
long up 45-00:00:0 3 mix lyowork[024-026]
long up 45-00:00:0 22 alloc lyowork[004-023,027-028]
gpu up 1-00:00:00 1 idle lyowork029
Where the node STATE means :
- idle : The worker nodes list are fully available to run new jobs
- mix : There are already some jobs running on these worker nodes, they are not fully booked, they still can run some new jobs
- alloc : The worker nodes list are fully booked, they can't run new jobs at this time
- drain or drng : The worker nodes list are in maintenance mode, they can't run new jobs at this time
Where for now the queues (called partitions) are :
| Partition name | Comment | Wall clock CPU time limit | Default RAM allocated (per core) | Maximum RAM allocated (per core) |
|---|---|---|---|---|
| normal | default partition | 24H (1 day) | 2 GB | 3 GB |
| long | for long jobs | 30 days | 2 GB | no limit |
| gpu | for jobs to be executed on the GPU server | 24H (1 day) | 2 GB | no limit |
| test | for short test jobs | 1H | 2 GB | 3 GB |
The Default RAM allocated and Maximum RAM allocated are given per thread (logical core)
if you don't specify any partition in the sbatch or srun commands (with -p or --partition option), your job will be launched in the normal partition.
The long partition is very relax at the moment to be able to fit any needs, but it will probably be refined when needed in the future
The command scontrol show partition normal gives for example the extended information about all the specifications of the normal partition (wall clock CPU time limit, default RAM allocated by core, the worker nodes list in this partition...).
Some examples
Run a command (here the hostname command) on 5 nodes:
srun --nodes=5 hostname
lyowork017.in2p3.fr
lyowork015.in2p3.fr
lyowork016.in2p3.fr
lyowork014.in2p3.fr
lyowork013.in2p3.fr
Yeah that was fast. The command hostname was run on 5 different workers which are in the normal partition, each time printing the worker hostname.
If you want to run 100 times the same hostname command, do:
$ srun --ntasks=100 hostname
This command spreads jobs to available worker nodes, for this example :
$ srun --ntasks=100 hostname | sort | uniq -c
30 lyowork005.in2p3.fr
30 lyowork006.in2p3.fr
30 lyowork007.in2p3.fr
10 lyowork008.in2p3.fr
Slurm has spread the 100 tasks to some workers (up to the maximum number of cores available of each worker).
A first batch script
You can submit a batch script to the SLURM batch system only from a shared filesystem (/home, /group, /gridgroup, but not from /scratch which is local to each server).
Here is an example batch script: submit.sh.
First, have a look in this script and edit this line with your email address:
#SBATCH --mail-user=d.pugnere@ip2i.in2p3.fr
You also see :
#SBATCH --output=submit.out
#SBATCH --error=submit.err
which redirects STDOUT (standard output) to the file submit.out and STDERR (standard error) to the file submit.err
You also see :
#SBATCH --ntasks=1
which is the default (1), you can delete this line if you don't need more
You also see :
#SBATCH --time=1:00:00 # means 1h 00m 00s wall clock time
which fixes the maximum time for this job, it will be killed by SLURM if it runs longer. For the default queue, the maximum running time is 24H wall clock time. The # character is for comments to be added.
You also see :
#SBATCH --mem-per-cpu=1G # I need not more than 1GB RAM
Which fixes the maximum usable RAM per CPU (per core). Without specifying this parameter, the job will herit the default and maximum RAM allocations per core defined by the partition on which it will be launched.
Currently, for the normal partition, the default RAM allocation per core is 2 GB and maximum RAM allocation is 3 GB.
If you need more RAM for your job, you can specify the ammount of RAM you need, up to the largest RAM available of the worker node on which the job will be executed.
For example :
#SBATCH --mem=50G
In this exemple, the SLURM scheduler will allocate a worker with enough RAM and reserve 50G of RAM for this job. Please that when you book more that the 3GB max per core, the SLURM batch system will allocate RAM from another core from the same worker node, i.e. if you book 9GB RAM, it will allocate 3 cores to you job even if you don't use the extra cores.
Then, submit and check the queue:
$ sbatch submit.sh
$ squeue
The output is created in submit.out file, as configured in the script. If any output error, it will be written to the submit.err file.
Interactive login on worker nodes
You can access worker nodes to test your jobs prior to send them to the batch system, you can also launch computation using this system.
The Interactive login on worker nodes is working like this in detail : When you launch an interactive login, the batch system books the CPUs you need for you and spawn a shell on one of the worker node available and allow you to execute your work.
For example : you need to log into a worker with defaults values (i.e. queue "normal" with 1 core, 24H CPU time and 2GB RAM per core) :
[pugnere@lyoui3:~/]$ srun --pty bash
[pugnere@lyowork005:~/]$ env|grep -E "SLURM_JOB_NODELIST|SLURM_JOB_NUM_NODES|SLURM_JOB_CPUS_PER_NODE"
SLURM_JOB_NODELIST=lyowork005
SLURM_JOB_CPUS_PER_NODE=1
SLURM_JOB_NUM_NODES=1
[pugnere@lyowork005:~/]$ exit
exit
[pugnere@lyoui3:~/]$
This previous command create a job in the batch system (you can see it with the squeue command), and then spawn a shell on the first idle worker node lyowork005. One CPU has been booked for you for 24H or until you exit this shell.
If you have a multithreaded process on only one worker node (posix threads or OpenMP for example) and if you need 16 cores :
[pugnere@lyoui3:~/]$ srun -c16 --pty bash
[pugnere@lyowork005:~/]$ env|grep -E "SLURM_JOB_NODELIST|SLURM_JOB_NUM_NODES|SLURM_JOB_CPUS_PER_NODE"
SLURM_JOB_NODELIST=lyowork005
SLURM_JOB_CPUS_PER_NODE=16
SLURM_JOB_NUM_NODES=1
But, if you need a worker node with 64 cores (posix threads or OpenMP) and there isn't a machine with this requirement in the batch system, you'll have an error :
[pugnere@lyoui3:~/]$ srun -c64 --pty bash
srun: error: CPU count per node can not be satisfied
srun: error: Unable to allocate resources: Requested node configuration is not available
If you want that your job will use all the RAM of one of the worker nodes, you can launch srun and specify the --exclusive or --mem=0 options, for example :
Whole CPUs worker node, 2GB RAM / slot :
[pugnere@lyoui3:~] $ srun --exclusive --pty bash
[pugnere@lyowork035:~] $ scontrol show job $SLURM_JOB_ID |grep -E "NumCPUs|mem="
NumNodes=1 NumCPUs=64 NumTasks=1 CPUs/Task=1 ReqB:S:C:T=0:0:*:*
TRES=cpu=64,mem=128G,node=1,billing=64
[pugnere@lyowork035:~] $ exit
Whole CPU's worker node, whole worker node's RAM :
[pugnere@lyoui3:~] $ srun --exclusive --mem=0 --pty bash
[pugnere@lyowork035:~] $ scontrol show job $SLURM_JOB_ID |grep -E "NumCPUs|mem="
NumNodes=1 NumCPUs=47 NumTasks=1 CPUs/Task=1 ReqB:S:C:T=0:0:*:*
TRES=cpu=47,mem=189745M,node=1,billing=47
[pugnere@lyowork035:~] $ exit
If you want an interactive login into a specific worker node, you should specify it in the --nodelist= option :
[pugnere@lyoui3:~] $ srun --nodelist=lyowork036--pty bash
[pugnere@lyowork036:~] $ exit
Interactive login on worker nodes with an X11 display
Now available and simple, just add the --x11 option :
[pugnere@lyoui3:~/]$ srun --pty --x11 xclock
or
[pugnere@lyoui3:~/]$ srun --pty --x11 bash
[pugnere@lyowork012:~/]$ xclock
Attaching to a running job
It is possible to connect to the node running a job and execute new processes there. You might want to do this for troubleshooting or to monitor the progress of one of your jobs.
Suppose you want to run the utility nvidia-smi to monitor GPU usage on a node where you have a job running. The following command runs watch on the node assigned to the given job, which in turn runs nvidia-smi every 30 seconds, displaying the output on your terminal.
[pugnere@lyoui3:~/]$ srun --jobid 123456 --pty watch -n 30 nvidia-smi
It is possible to launch multiple monitoring commands using tmux. The following command launches htop and nvidia-smi in separate panes to monitor the activity on a node assigned to the given job.
[pugnere@lyoui3:~/]$ srun --jobid 123456 --pty tmux new-session -d 'htop -u $USER' \; split-window -h 'watch nvidia-smi' \; attach
How to process a bunch of files, with only 1 job launching as sub-tasks as files ?
By default, 1 batch job = 1 task.
But you can also have 1 job that will have many independant tasks, and even have some tasks which depends upon others. Let see one simple example when you need to process many files the same way with the same executable.
There are 2 main ways to do that :
- the first trivial way to do it is to launch a job for each file. In this case, you will have as many jobs as files to process. This is not the best way to do this kind of processing because it creates a lot of independant jobs, many output files.
- The better way to do this is to launch only 1 job and launch tasks inside this job to process your files. In this case, you'll have only 1 job and as many tasks as files to process inside this job. But the trick is that you can have your tasks running in parallel using the
--ntasksparameter.
Example, if we have a directory in which we have 1000 files (or whatever the number), we need to process each file with the same executable per file, with for example 200 sub-jobs max running at a time on the cluster. This is simple, we create 1 only job with 200 (independant and parallel) sub tasks :
Exemple for a shell job-process-files.sh :
#!/bin/bash
#
#SBATCH --ntasks=200
#SBATCH --spread-job
DIR=/home/infor/pugnere/encours/slurm/job-tasks-dir
for file in $DIR/*
do
srun -n1 ./job-process-files-task.sh $file &
done
# Waiting all the sub-tasks to finish before continuing
wait
echo "End of the job"
The loop for will take each file in the $DIR directory and lauch as sub-tasks (srun command) as the number of files.
Note the -n1 and the & (background) : srun will allocate 1 CPU on a node and launch the sub-task on it. The -n1 parameter is important, it specifies that this is 1 task, also the & which spawn the task. If each of your tasks need more than 1 CPU (for multi-threads tasks), you can also specify the --cpus-per-task=x parameter on the srun command. Note also the wait command which waits that all the sub-tasks to finish before reaching the end of the job's script.
Now, for the script job-process-files-task.sh which will process each of your files, this can be a simple :
#!/bin/bash
# the file name to process will be given as a parameter
echo sub-task $SLURM_STEP_ID
./process_file $1
You just have to make them executable :
$ chmod +x job-process-files.sh job-process-files-task.sh
and then send it to the batch system :
$ sbatch job-process-files.sh
and then run :
$ squeue
JOBID PARTITION NAME USER ST TIME NODES NODELIST(REASON)
4102 normal job-proc pugnere R 0:08 7 lyowork[013-019]
We see 1 job which is running sub-tasks on several workers.
Please, choose the good number of tasks : Number of sub-tasks not to high if the cluster is already heavily used, because your job will have to wait to have enough CPU available before enter in RUNNING state.
Please note that when you have I/O intensive processing, it should be the case if you process a large number of files, or large files, you will get a huge speedup using the --spread-job parameter to sbatch. In this case, SLURM will spread the job allocation over as many nodes as possible and attempt to evenly distribute tasks across the allocated nodes. You will benefit of the cumulated network or disk bandwidth of each node.
Informations on jobs arrays
Job arrays offer a very simple way to submit a large number of independant jobs. They can typically be used to apply the same program to different input data.
Submitting a job array is done simply by adding the option
--array=<indices>
or adding in the header or the job's script :
#SBATCH --array=0-9 # create an array of 10 jobs (indices from 0 to 9)
A job array is made up of a main job (the array) and tasks (the jobs contained in the array). SLURM assigns a number to a job in the array, consisting of the number of the main job and its index: <SLURM_ARRAY_JOB_ID >_<SLURM_ARRAY_TASK_ID>.
The following environment variables are available:
- SLURM_ARRAY_JOB_ID: Number of the main job.
- SLURM_ARRAY_TASK_ID : index of the task in the table
- SLURM_ARRAY_TASK_COUNT: total number of jobs in a job array
- SLURM_ARRAY_TASK_MAX : max index of the job table
- SLURM_ARRAY_TASK_MIN : min index of the job table
Create parallel multi CPU jobs (OpenMP or MPI)
Parallel (or multi) CPU jobs workloads are executables which use more than 1 core to do calculations, they enables the creation of concurrent process flows. It works well on multi-processor or multi-core systems, where the process flow may be scheduled to execute on another processor, increasing speed through parallel or distributed processing.
There are 2 kinds of parallel workloads :
- OpenMP (POSIX Threads, commonly known as pthreads) is a library which allow processes to use several cores which can only be executed inside one worker node. It can't be spreaded over worker nodes, pthreads can't comunicate outside the worker nodes. It can use as many cores available in a worker node.
- MPI : Message Passing Interface (MPI) is a standardized and portable message-passing standard designed to function on parallel computing architectures. This library which allow processes to use several cores which can be executed on more than one worker node, the library can comunicate outside the worker nodes. It can use as many cores available in a worker node as wall as many worker nodes as necessary.
To launch OpenMP or MPI jobs, create scripts with one or a combination of these headers :
#!/bin/bash
# -n=x | --ntasks=x : total number of cores (for MPI, can be spreaded on several worker nodes)
#SBATCH --ntasks=128
# -c=x | --cpus-per-task=x : number of cores on ONE worker node (for posix threads or OpenMP)
#SBATCH --cpus-per-task=16
# -N=x | --nodes=x : number of worker nodes if you need to spread your work over some worker nodes
#SBATCH --nodes=3
...
Create OpenMP (Posix Threads) jobs
The OpenMP (Posix Threads) executables also need to know how many threads (so CPU cores) they can use, the default is 1, but SLURM put some information in the ENVIRONMENT so the OpenMP (Posix Threads) executables can read this indication from ENVIRONMENT variables. We can explicitly declare the number of threads the executable can use, we get the values from SLURM env variables ($SLURM_NTASKS, and indirectly $SLURM_TASKS_PER_NODE).
For example, for a pure OpenMP job :
#!/bin/bash
# -c=x | --cpus-per-task=x : number of cores on ONE worker node (for posix threads or OpenMP)
#SBATCH --cpus-per-task=16
# we must instantiate the control variable OMP_NUM_THREADS
# which tells the binary the maximum number of threads it can use
# this variable must correspond to the specification of the slurm job
# taken from the --cpus-per-task parameter
# this aprameter is available in SLURM_CPUS_PER_TASK or $SLURM_NTASKS environemnt variable
# example :
# export OMP_NUM_THREADS=$SLURM_NTASKS
#
# or for a mixed MPI / OpenMP job :
# export OMP_NUM_THREADS=`expr $SLURM_JOB_CPUS_PER_NODE / $SLURM_TASKS_PER_NODE`
export OMP_NUM_THREADS=$SLURM_CPUS_PER_TASK
./my-openmp-job
...
Create MPI jobs
The interest of the MPI library is to be able to use several nodes in parallel. The following script shows how to launch 16 processes in parallel distributed on 4 nodes. It requires the creation of a machine file which contains the list of machines to be used by MPI with the number of processes to launch on each one.
$ cat job-mpi.machines
lyowork001 slots=4
lyowork002 slots=4
lyowork003 slots=4
lyowork004 slots=4
The options given to SLURM must be consistent with the information contained in the machine file :
$ cat job-mpi.sh
#!/bin/bash
#SBATCH --ntasks=16
#SBATCH --ntasks-per-core=1
#SBATCH --job-name=testMPI
#SBATCH --nodelist=lyowork[007-008]
# the file job-mpi.machines should contain the machine list to use
# it should be the same as the value of the --nodelist option
module load mpi/mpich-3.2-x86_64
mpiexec -machinefile job-mpi.machines ./job-mpi
Then send the job :
$ sbatch job-mpi.sh
Some other reservations
Now, you have MPI jobs, or hybrid jobs (MPI + OpenMP) to test which can be spreaded over multiple worker nodes, you can specify the total number of cores you need, even if this larger than the largest worker node, in this case, you can use the salloc command :
[pugnere@lyoui3:~/]$ salloc -n 128
salloc: Granted job allocation 4170
salloc: Waiting for resource configuration
salloc: Nodes lyowork[013-017] are ready for job
...
[pugnere@lyoui3:~/]$ env|grep -E "SLURM_JOB_NODELIST|SLURM_JOB_NUM_NODES|SLURM_JOB_CPUS_PER_NODE|SLURM_NPROCS"
SLURM_NPROCS=128
SLURM_JOB_NODELIST=lyowork[013-017]
SLURM_JOB_CPUS_PER_NODE=30(x4),8
SLURM_JOB_NUM_NODES=5
[pugnere@lyoui3:~/]$ module load mpi/mpich-3.2-x86_64
[pugnere@lyoui3:~/]$ mpirun ./my_mpi_job
[pugnere@lyoui3:~/]$ exit
In this previous example, the worker nodes lyowork013 to lyowork017 are booked with 30 * 4 + 8 CPU booked for you and when you'll launch your mpi job, it will use this reservation. This is useful for testing purpose.
Some other example of SLURM batch scripts
Here are some other job script examples :
- job-redir.sh : Example of how to redirect STDOUT and STDERR to files
- job-partition.sh : Example of how to send a job to the
longpartition - job-mail.sh : Example of how to send a mail at the end of the job
- job-ram.sh : Example of a how to book enough RAM to the job
- job-tasks.sh and job-tasks-exec.sh : Example of a how to launch sub-tasks
- job-tasks-spread.sh and job-tasks-exec.sh : Example of a how to process a large number of files
- job-openmp.sh and C source job-openmp.c : Example of a script which compile and run an OpenMP program which can use 10 threads (CPU cores)
- job-mpi.sh and C source job-mpi.c : Example of a script which compile and run en MPI program which can use 100 CPUs (CPU cores from 1 or several nodes)
sbatch Options
The sbatch command has these options :
| Option | Long Option | Comment |
|---|---|---|
| -c, | --cpus-per-task= |
number of CPU asked for 1 task |
| -n, | --ntasks= |
number of tasks for a job |
| --ntasks-per-node= |
number of tasks asked for each node | |
| -N, | --nodes= |
number of nodes asked (N = min[-max]), N = 1 by default |
| -C, | --constraint= |
//features// asked (multiple features may be specified, in this case, separate them by &) |
| --mem= |
minimal RAM size (in MB) asked per job | |
| --mincpus= |
minimal numbrer of CPU (threads) per node = number of threads | |
| -w, | --nodelist= |
ask for thises nodes (separated by ,), this list should contains enought node asked bu the option --nodes |
| -x, | --exclude= |
exclude all the nodes specified for the execution of the job (separated by ,) |
| -a, | --array= |
indexes value for an array of jobs |
| -e, | --error= |
file name of the job's error output |
| -o, | --output= |
file name of the standard output |
| -I, | --immediate | the job is killed if the ressources are not immediatly available |
| -J, | --job-name= |
give it's name to the job |
| --mail-type= |
notification by mail, type = BEGIN, END, FAIL, REQUEUE, ALL, STAGE_OUT, TIME_LIMIT_90, TIME_LIMIT | |
| --mail-user= |
mail adress to send notifications | |
| -t, | --time= | maximum time allocated to the job (format = m:s ou h:m:s ou j-h:m:s) |
Control your jobs
To see all informations about a RUNNING job :
$ scontrol show job <job_id>
To cancel a job :
$ scancel <job_id>
Waiting time
Some commands to show jobs in the queue :
squeuedisplays current and pending jobs in order of priority.squeue --priority --sort=-p,i --states=PDList of pending jobs in the same order considered for scheduling by Slurmsqueue -u <user>displays current and pending jobs for the user usersqueue -p <partname>displays current and pending jobs for the requested partitionsqueue -i <sec>refreshes the list of running jobs every sec
sprio gives priority to pending jobs (the highest priority jobs have the highest priority)
Deleting a job
scancel <jobID>delete job(running or pending) scancel -u <user>deletes jobs from user(running or pending)
Status of jobs
Jobs typically pass through several states in the course of their execution. The most simplified workflow of jobs states is :
- PENDING -> RUNNING -> COMPLETED (if job exit status == 0)
- PENDING -> RUNNING -> FAILED (if job exit status != 0)
The typical states are PENDING, RUNNING, SUSPENDED, COMPLETING, and COMPLETED. An explanation of each state follows, the possible states are :
| Abbreviation | Job state | Explanation |
|---|---|---|
| BF | BOOT_FAIL | Job terminated due to launch failure, typically due to a hardware failure (e.g. unable to boot the node or block and the job can not be requeued). |
| CA | CANCELLED | Job was explicitly cancelled by the user or system administrator. The job may or may not have been initiated. |
| CD | COMPLETED | Job has terminated all processes on all nodes with an exit code of zero. |
| CF | CONFIGURING | Job has been allocated resources, but are waiting for them to become ready for use (e.g. booting). |
| CG | COMPLETING | Job is in the process of completing. Some processes on some nodes may still be active. |
| DL | DEADLINE | Job terminated on deadline. |
| F | FAILED | Job terminated with non-zero exit code or other failure condition. |
| NF | NODE_FAIL | Job terminated due to failure of one or more allocated nodes. |
| OOM | OUT_OF_MEMORY | Job experienced out of memory error. |
| PD | PENDING | Job is awaiting resource allocation. |
| PR | PREEMPTED | Job terminated due to preemption. |
| R | RUNNING | Job currently has an allocation. |
| RD | RESV_DEL_HOLD | Job is held. |
| RF | REQUEUE_FED | Job is being requeued by a federation. |
| RH | REQUEUE_HOLD | Held job is being requeued. |
| RF | REQUEUE_FED | Job is being requeued by a federation. |
| RH | REQUEUE_HOLD | Held job is being requeued. |
| RQ | REQUEUED | Completing job is being requeued. |
| RS | RESIZING | Job is about to change size. |
| RV | REVOKED | Sibling was removed from cluster due to other cluster starting the job. |
| SI | SIGNALING | Job is being signaled. |
| SE | SPECIAL_EXIT | The job was requeued in a special state. This state can be set by users, typically in EpilogSlurmctld, if the job has terminated with a particular exit value. |
| SO | STAGE_OUT | Job is staging out files. |
| ST | STOPPED | Job has an allocation, but execution has been stopped with SIGSTOP signal. CPUS have been retained by this job. |
| S | SUSPENDED | Job has an allocation, but execution has been suspended and CPUs have been released for other jobs. |
| TO | TIMEOUT | Job terminated upon reaching its time limit. |
Fair share information
Fairshare allows past resource utilization information to be taken into account into job feasibility and priority decisions to ensure a fair allocation of the computational resources between the all IP2I's users. Every participating research group is entitled for the same amount of resources. This entitlement is shared between all accounts and its members using that accounts.
The fair-share system is designed to encourage users to balance their use of resources over time and de-prioritize users/accounts with over average usage. Thus no user group/research group gets the opportunity to over-dominate on the systems, while others request resources. Fair-share is the largest factor in determining priority, but not the only one.
One parameters to be taken into account : There is an 1 week half-life, this is the time, of which the resource consumption is taken into account for the Fairshare Algorithm.
To see your fair share and all IP2I's groups :
$ sshare
To see all fair shares of all IP2I's groups :
$ sshare -a
To see fair shares of all users of your group :
$ sshare --accounts=ACCOUNT_GROUP
Accounting information
When a job is finished, we dont see it anymore with the squeue command, but it state and all accounting information are archived in an accounting database for several months. This accounting information can be retreived with the sacct command.
sacct displays the status of the user's jobs whether they are in progress or already completed.
Invocation exemple :
sacct --format=JobID,partition,state,elapsed,AveCPU,MaxRss,MaxVMSize,nnodes,ncpus
Efficiency of your jobs
To see efficiency of your job (CPU part versus waiting for I/O ) :
seff JOB_ID